CRYSOFT – IC BARI
DESCRIPTION:
Development of computational methodologies for the structural solution of molecules of biological or biotechnological interest with different complexity by means of crystallography (CrySoft)
The crystallographic methodologies have a foundamental importance to study the condensed matter. They allow to understand the crystal structure starting from diffraction data. The physical, chemical and biological properties of different samples strongly depend on the relative atomic positions in the molecules.
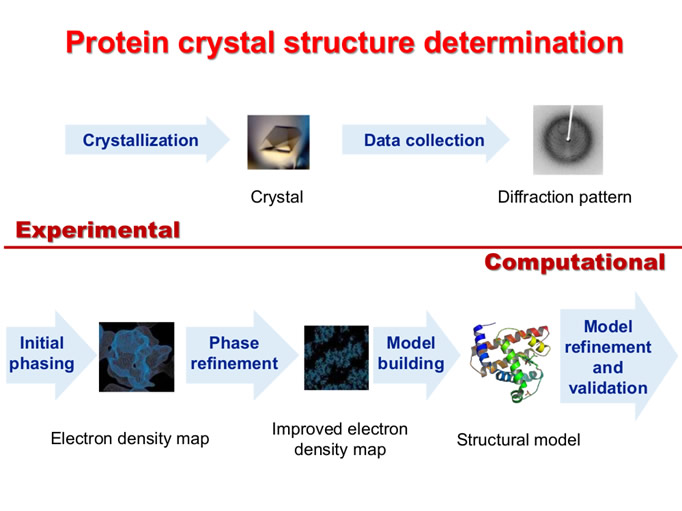
The activity of the CrySoft research group is located between the chemistry and the structural biology area; it is devoted to:
• development of innovative crystallographic methodologies for the ab-initio and not ab-initio crystal structure solution of different complexity materials, using single crystal diffraction data (RX or electrons),
• implementation of new theories and algorithms in crystallographic software,
• application of these new procedures to experimental data (i.e structural determination of drugs or material of biological and technological interest)
This activity is totally consistent with the tradition of the Institute of Crystallography, scientific leader at international level for the developed theories and related software.
Within the framework of the studies on small/medium size molecules (i.e. drugs or ligands) the activity has been focused on the continuous enhancement of the algorithms devoted to ab-initio phasing in reciprocal space (Direct Methods) and on improvement of the phase extension and refinement in direct space updating the algorithms involved in direct space refinement (DSR) procedure.
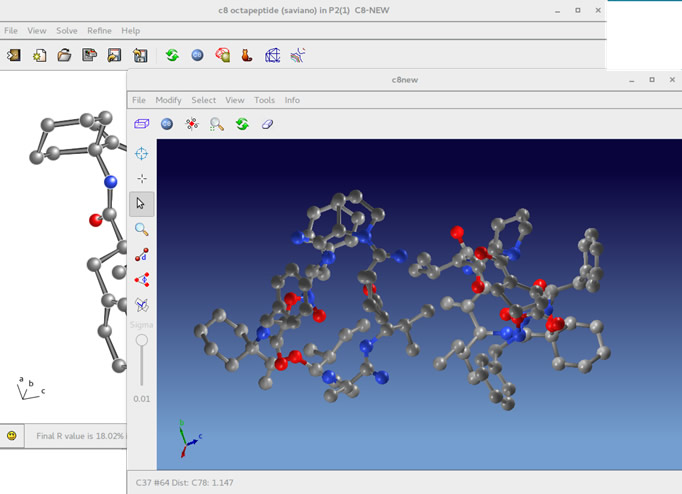
Default automatic solution of a small molecule
About the structural determination of macromolecules, often characterized by incomplete diffraction data at low resolution, most of the time it is problematic and no completely automatized. The researches in this field are related to study new procedures able to facilitate the crystal structure solution of proteins introducing, in the different techniques used (ab-initio, MAD/SAD, MIR/SIR, Molecular Replacement, in synergy with the Phantom Derivative method) the innovative procedures deriving from the theoretical studies established in IC.
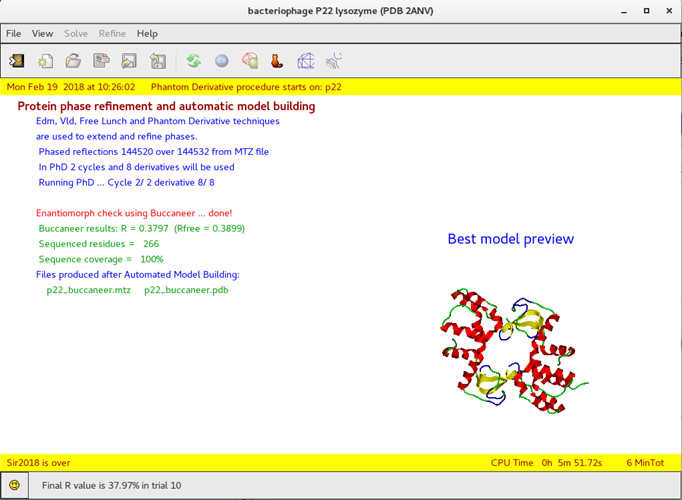
Default automatic solution of a protein
The different algorithms devoted to the crystal structure solution (using single crystal data) are implemented in the programs Sir2018 and Il Milione, in order to offer, to the international community, an effective, flexible and performing software easy to use.
The software is free for academic researchers and it is used in many labs all over the world.
Contact: GIOVANNI LUCA CASCARANO – gianluca.cascaranoATic.cnr.it
INVOLVED STAFF:
Staff:
Benedetta Carrozzini, Rocco Caliandro, Giovanni Luca Cascarano
Coworkers:
Carmelo Giacovazzo, Maria Cristina Burla, Giampiero Polidori
FACILITIES AND LABORATORIES:
(None at the Moment)
GROUP RELATED RESEARCH PROJECTS:
(None at the Moment)
GROUP RELATED PUBLICATIONS :
- Burla M.C., Carrozzini B., Cascarano G.L., Giacovazzo C. & Polidori G. (2017) – Solving proteins at non-atomic resolution by Direct Methods: update – Jour. Appl. Cryst., 50, 1048-1055.
- Burla M.C., Carrozzini B., Cascarano G.L., Giacovazzo C. & Polidori G. (2015) – Solving protein at non-atomic resolution by Direct Methods- Jour. Appl. Cryst., 48, 1692-1698.
- Burla M.C., Carrozzini B., Cascarano G.L., Giacovazzo C. & Polidori G. (2015) – Refining a model electron-density map via the Phantom Derivative method – Acta Cryst., D71, 1864-1871.
- Burla M.C., Caliandro R., Carrozzini B., Cascarano G.L., Cuocci C., Giacovazzo C., Mallamo M., Mazzone A. & Polidori G. (2015) – Crystal structure determination and refinement via SIR2014- Jour. Appl. Cryst., 48, 306-309.
- Caliandro R., Carrozzini B., Cascarano G.L., Comunale G., Giacovazzo C. & Mazzone A. (2014) – Protein phasing at non-atomic resolution by combining Patterson and VLD techniques. – Acta Cryst., D70,